Command line reference
Specify the input file in FASTA format containing one or more RNA sequences as well as the path where the output files will be created (the folder will be created if it does not exist).
r2dt.py draw <input.fasta> <output_folder>
For example:
r2dt.py draw examples/examples.fasta temp/examples
R2DT will automatically select the best matching template and visualise the secondary structures.
Output files
r2dt.py draw produces a folder called results with the following subfolders:
svg: RNA secondary structure diagrams in SVG formatfasta: input sequences and their secondary structure in dot-bracket notationtsv: a filemetadata.tsvlisting sequence ids, matching templates, and template sourcesthumbnail: secondary structure diagrams displayed as outlines in SVG formatjson: RNA secondary structure and its layout described using RNA 2D JSON Schema
Manually selecting template type
If the RNA type of the input sequences is known in advance, it is possible to bypass the classification steps and achieve faster performance.
CRW templates (5S and SSU rRNA)
r2dt.py crw draw examples/crw-examples.fasta temp/crw-examples
RiboVision LSU and SSU rRNA templates
r2dt.py ribovision draw_lsu examples/lsu-examples.fasta temp/lsu-examples r2dt.py ribovision draw_ssu examples/ribovision-ssu-examples.fasta temp/ssu-examples
Rfam families
r2dt.py rfam draw RF00162 examples/RF00162.example.fasta temp/rfam-example
RNAse P
r2dt.py rnasep draw examples/rnasep.fasta temp/rnasep-example
tRNAs (using GtRNAdb templates)
# for tRNAs, provide domain and isotype (if known), or use tRNAScan-SE to classify r2dt.py gtrnadb draw examples/gtrnadb.E_Thr.fasta temp/gtrnadb r2dt.py gtrnadb draw examples/gtrnadb.E_Thr.fasta temp/gtrnadb --domain E --isotype Thr
Manually selecting specific template
It is possible to select a specific template and skip the classification step altogether.
Get a list of all available templates and copy the template id:
r2dt.py list-models
In addition, all models are listed in the file models.json.
Specify the template (for example,
RNAseP_a_P_furiosus_JB):r2dt.py draw --force_template <template_id> <input_fasta> <output_folder>
For example:
r2dt.py draw --force_template RNAseP_a_P_furiosus_JB examples/force/URS0001BC2932_272844.fasta temp/example
Constraint-based folding for insertions
If a structure contains insertions relative to the R2DT template files, they are shown as large unstructured loops. Insertions larger than 100 nucleotides are replaced with a placeholder element (XXXX characters) to keep the rest of the diagram legible.
The --constraint flag allows de-novo prediction of the insertion structure using the RNAfold algorithm. There are currently three constraint folding modes available. R2DT will automatically predict which folding mode is best for a given molecule, but the mode can also be manually overridden using the –fold_type parameter. There are three options for fold_type.
Let R2DT pick a fold_type
r2dt.py draw --constraint <input_fasta> <output_folder>
Fold insertions (along with adjacent unpaired nucleotides) one at a time. Recommended for large RNAs.
r2dt.py draw --constraint --fold_type insertions_only <input_fasta> <output_folder>
Run entire molecule through RNAfold at once. Base pairs predicted from the template are used as constraints for prediction.
r2dt.py draw --constraint --fold_type full_molecule <input_fasta> <output_folder>
Run entire molecule through RNAfold at once. Both conserved single-stranded regions and base pairs predicted from the template are used as constraints for prediction.
r2dt.py draw --constraint --fold_type all_constraints_enforced <input_fasta> <output_folder>
Prevent certain nucleotides from base pairing. This will only work for base pairs that are de-novo predicted. The exclusion file should contain a string the same length as the input sequence composed of ‘.’s and ‘x’s. Positions with ‘.’s are allowed to base pair, positions with ‘x’s are not. Example string:
xxxx..............xx..............x............xxr2dt.py draw --constraint --exclusion <exclusion_file> <input_fasta> <output_folder>
Template-free visualisation
Starting with version 1.4, it is possible to visualise a sequence and its secondary structure using a layout generated by R2R. This functionality is useful as a starting point when generating new templates or in cases when the R2DT template library does not yet have a template for a certain RNA.
Example input found in examples/template-free.fasta (pseudoknots can be specified in the Aa, Bb notation):
>3SKZ_B
GGCCUUAUACAGGGUAGCAUAAUGGGCUACUGACCCCGCCUUCAAACCUAUUUGGAGACUAUAAGGUC
.((((((((A..((((((.....BB))))))(.....a)(((((((bb..)))))))..)))))))).
Example command:
r2dt.py templatefree examples/template-free.fasta temp/template-free-example
Output diagram:
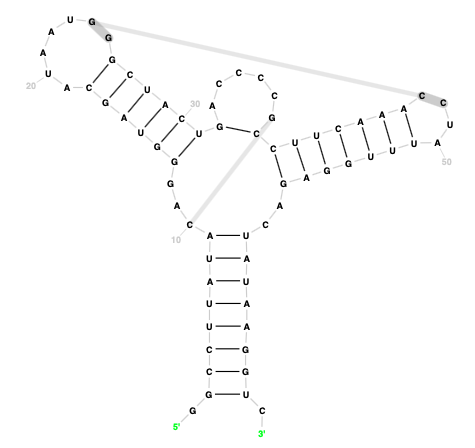
The output files are organised in the standard way and include SVG and RNA 2D JSON Schema files. The JSON file can be uploaded in the interactive editor for further manual editing, if necessary.
Skipping ribovore filters
In some cases R2DT may not generate a diagram for a sequence because ribovore detects one or more unexpected features, such as having hits on both strands or having too many hits in the same sequence. You can use --skip_ribovore_filters to ignore these warnings and attempt to generate a secondary structure diagram anyway.
For example, the following command will produce no results because the sequence is close to a palindrome:
r2dt.py draw examples/ribovore-qc-example.fasta temp/examples
However, the following command generates a valid diagram:
{kind=link}
r2dt.py draw --skip_ribovore_filters examples/ribovore-filters.fasta temp/examples
Please note that this option should be used with caution as sequences with unexpected features often result in poor diagrams.
Other useful commands
Print R2DT version
r2dt.py versionClassify example sequences using Ribotyper
perl /rna/ribovore/ribotyper.pl -i data/cms/crw/modelinfo.txt -f examples/pdb.fasta temp/ribotyper-test
Generate covariance models and modelinfo files
python3 utils/generate_cm_library.py r2dt.py generatemodelinfo <path to covariance models>
Precompute template library locally (may take several hours):
r2dt.py setupRun R2DT with Singularity
singularity exec --bind <path_to_cms>:/rna/r2dt/data/cms r2dt r2dt.py draw sequence.fasta output
Convert a SVG diagram to a JSON file containing the paths per nucleotide and an ordinal numbering. Note that this assumes that the input pdb id is formatted like:
<PDB>_<Entity>_<chain>, ie1S72_1_0.svg2json.py <pdb-id> diagram.svg <pdb-id>.json